Ryan Kingsbury
Incoming Assistant Professor (2023)
Department of Civil and Environmental Engineering, Princeton University
Andlinger Center for Energy and the Environment, Princeton University
Biography
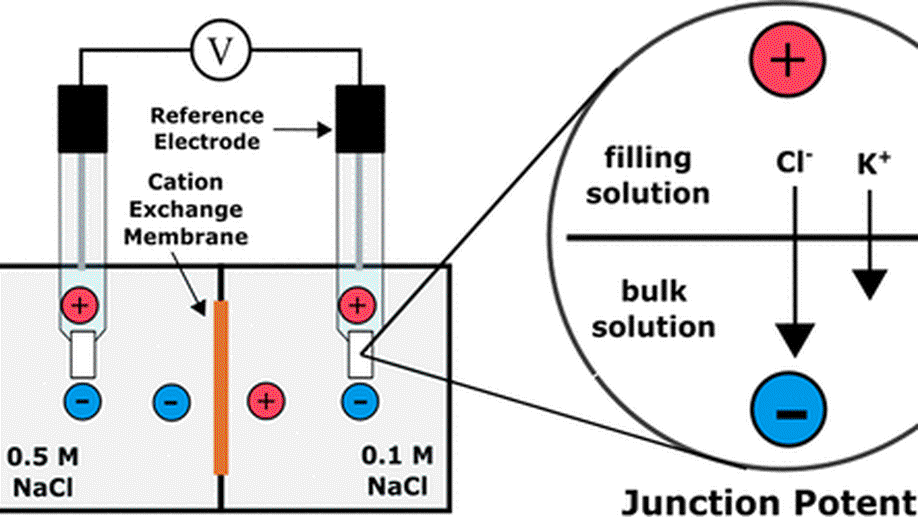
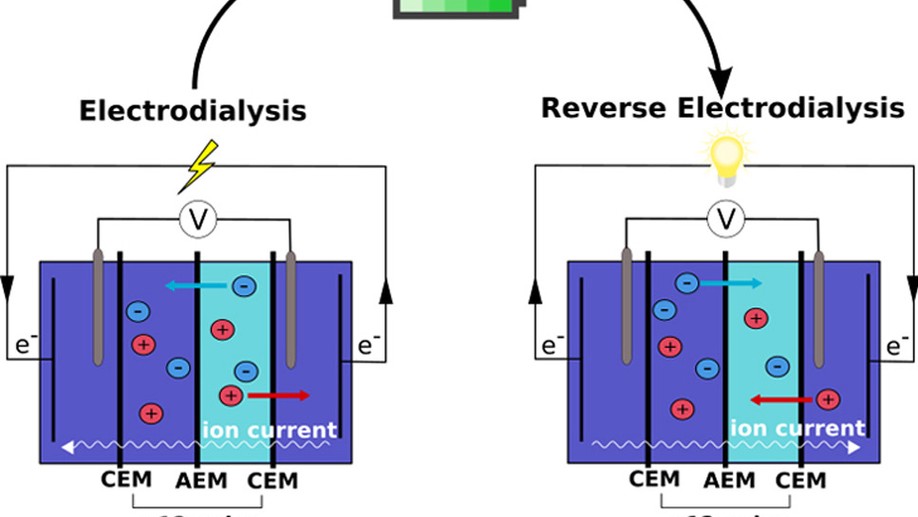
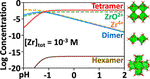
My research aims to accelerate the development of electrochemical technologies for clean water and clean energy production by advancing fundamental understanding of ion-selective materials such as membranes and electrodes. These materials preferentially absorb or transport charged particles like Lithium or Sodium ions and are found in numerous environmental technologies including water desalination systems, fuel cells, electrolyzers, and flow batteries. I seek to develop a molecular-level understanding of the factors that make these materials selective to certain ions, and use that knowledge to engineer higher-performing materials. My multi-scale research approach integrates electrochemical and thermodynamic materials characterization methods, first principles simulations, and device testing to understand how materials behave under a wide variety of conditions.
Interests
- Membrane separation processes
- Ion-selective materials
- Electrochemical technologies
- Computational materials discovery
Education
PhD in Environmental Sciences and Engineering, 2019
University of North Carolina at Chapel Hill
Master of Science in Environmental Engineering, 2010
University of North Carolina at Chapel Hill
B.S. in Civil and Environmental Engineering, 2007
University of Texas at Austin
B.A. in Plan II, 2007
University of Texas at Austin